Usage
Simple usage
The easiest form of usage is:
RnaChipIntegrator GENES PEAKS
where GENES and PEAKS are tab-delimited files containing
the gene and peak data respectively (see Input files for details
of these files).
This will produce two output files:
GENES_peak_centric.txt: reports the nearest genes for each peak (‘peak-centric’ analysis)
GENES_gene_centric.txt: reports the nearest peaks for each gene (‘gene-centric’ analysis)
In both cases the files will contain one peak/gene pair per line (see Output files for details of these files).
The program has various options that can be applied to control the analyses that are performed and the outputs from each run, as outlined in the following sections.
Specifying distance cutoff (--cutoff)
The --cutoff option specifies a maximum distance in bp that a
gene/peak pair can be apart and still be included in the analyses;
gene/peak pairs which are further apart than this distance will
not be reported.
For example:
RnaChipIntegrator --cutoff=130000 GENES PEAKS
Note
If a maximum cutoff distance is not explicitly specified then the default is 1000000 bp. Set the distance to 0 to turn off the cutoff limit and include all pairs regardless of distance.
Specifying how distances are measured between peaks and genes (--edge)
By default the distance between a peak and a gene is calculated as the distance from the gene TSS to the nearest peak edge, for example:
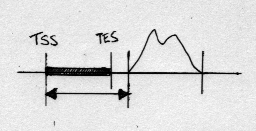
(This behaviour can be made explicit by specifying the
--edge=tss option.)
Alternatively distances can be calculated as the distance from
the gene TES (by specifying the --edge=tes), or as the shortest
distance between either of the peak edges to whichever of the TSS or
the TES of the gene is closest (by specifying the --edge=both
option).
For example:
RnaChipIntegrator --edge=both GENES PEAKS
For example for the same arrangement as above this would generate a much smaller closest distance:
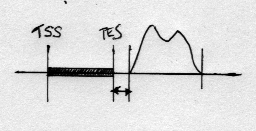
Note
Using --edge=both essentially makes the analyses
‘strand-agnostic’.
Only using differentially expressed genes (--only-DE)
If the input genes data contains a differential expression flag
(see ‘Genes’ data file) then this can be used in the analysis
by turning on the --only-DE option:
RnaChipIntegrator --only-DE GENES PEAKS
which will only included the flagged genes in the analyses.
Note
Without the --only-DE option, all genes will be used
regardless of the presence of a differential expression
flag.
Limiting the number of results to report (--number)
By default, all gene/peak pairs that are located within the specified cut-off distance (see Specifying distance cutoff (--cutoff)) will be reported in the output files.
To restrict the maximum number of pairs that are reported per gene
or peak use the --number to specify a limit. Even if more pairs
are found, only this number of pairs will be output.
Warning
Be aware that if used, this number limit is applied rigidly.
For example, even if the fourth and fifth gene/peak pairs both
have the same distance separation then using --number=4
will only include the first of these and reject the second.
Specifying the promoter region (--promoter_region)
As part of its peak-centric analyses, for each peak/gene pair
RnaChipIntegrator reports whether the peak overlaps the
promoter region of the gene.
By default, within the program the promoter region of a gene is defined as starting 1000 bp upstream of the gene TSS and ending 100 bp downstream of the TSS.
The --promoter_region option can be used to define a different
set of limits for this region, using the general format:
--promoter_region=UPSTREAM,DOWNSTREAM
For example:
--promoter_region=1500,200
would define a promoter region starting 1500 bp upstream of the TSS and ending 200 bp downstream.
Running either peak-centric or gene-centric analysis only (--analyses)
By default RnaChipIntegrator runs both peak-centric and
gene-centric analyses.
However it is possible to restrict the program to just one or
other of these, by using the --analyses option.
For example to run only the peak-centric analyses:
--analyses=peak_centric
Or, to run only the gene-centric analyses:
--analyses=gene_centric
The advantage of restricting the analyses is that it reduces the program run time, and limits the outputs to only those specifically requested.
Specifying multiple distance cutoffs (--cutoffs)
RnaChipIntegrator can peform its analyses over multiple cutoff
distances by using the --cutoffs option to supply a comma-separated
list of distances, for example:
RnaChipIntegrator --cutoffs=50000,100000,150000 GENES PEAKS
The selected analyses will be repeated for each of the specified cutoff distances, and the distance will be reported as an additional field for each gene/peak pair in the output files (see Additional fields for batch operation).
Note that --cutoffs is an alternative to the --cutoff option
and the two cannot be used together.
Note
This option can be used along with --peaks and
genes (see Specifying multiple peaks and/or genes files (--peaks and --genes)), to apply several
cutoff distances to analyses of multiple peaks and/or genes
files.
Specifying multiple peaks and/or genes files (--peaks and --genes)
In normal operation RnaChipIntegrator operates on a single pair
of files specifying the gene and peak data.
However it can also operate on multiple peaks and/or genes files
within a single run, by using the --peaks and --genes options.
For example, to analyse a pair of genes sets against the same set of peaks:
RnaChipIntegrator --genes GENES1 GENES2 --peak PEAKS
which would result in the program performing two analyses (i.e.
GENES1 versus PEAKS and GENES2 versus PEAKS).
Analysing several sets of peaks against a single set of genes would look like:
RnaChipIntegrator --genes GENES --peak PEAKS1 PEAKS2 PEAKS3
which would result in the program performing three analyses (i.e.
GENES versus PEAKS1, PEAKS2 and PEAKS3).
Analysing multiple sets of genes against multiple sets of peaks would look like:
RnaChipIntegrator --genes GENES1 GENES2 --peak PEAKS1 PEAKS2 PEAKS3
This would result in the program performing six analyses (i.e.
GENES1 versus PEAKS1, PEAKS2 and PEAKS3 then GENES2
versus the three peaks files).
Note that --peaks and --genes must always be used together,
and instead of specifying a single pair of files at the end of the
command line.
In all cases where there is more than one file then the name of the appropriate file(s) will be reported as an additional field for each gene/peak pair in the output files (see Additional fields for batch operation).
Note
These options can be used along with --cutoffs (see
Specifying multiple distance cutoffs (--cutoffs)), to repeat each set of
analyses at various cutoff distances.
Specifying multiple cores in batch modes (--nprocessors)
RnaChipIntegrator can use multiple cores in ‘batch’ modes (that
is, any run which performs more than one analysis because multiple
distance cutoffs and/or multiple peaks or genes files were specified
on the command line).
In these modes the number of cores to use can be supplied via
the --nprocessors option, for example:
RnaChipIntegrator --cutoffs=50000,100000,150000 --nprocessors=2 GENES PEAKS
Changing the output files and formats
There are a number of options to produce additional output files, and to modify the format and output content depending on requirements:
Using RnaChipIntegrator in Galaxy
In addition to the command-line version, we have also provided a tool
which allows RnaChipIntegrator to be run within the popular
Galaxy bioinformatics platform:
The tool can be installed into a local instance of Galaxy directly from the Galaxy Toolshed
See the documentation at http://getgalaxy.org/ on how to get a local Galaxy up and running, and how to install tools from the Toolshed.